食品中总砷测定前处理
食品中总砷测定前处理
摘要:
目的 探索试样前处理过程中,赶酸条件控制的优化,试验用容器具中被测元素本底残留对原子荧光光度法检测痕量砷的污染权重放大效应。
方法 采用湿消解法和微波消解法,在不同条件赶酸,用氢化物原子荧光光度法检测标准物质小麦粉中的砷含量,并与标准值比对。
结果 试样前处理赶酸是否规范,将直接影响检测结果的准确性。被测元素本底污染权重放大作用,使检测结果高出标准值可信范围。
结论 严格按照消解采用的酸的沸点控制赶酸温度,标准物质检测值在标准值可信范围。
关键词:食品;砷;荧光光度测定法;研究技术
在食品中砷的检测过程中,前处理是否彻底、酸度残留、实验器皿的污染程度会直接影响消解试液的上机检测结果。在日常检测工作中,有些细节往往被忽视,导致检测结果偏倚,作者就这些问题进行探索性研究。
1 材料与方法
1. 1 材料
标准物质小麦粉
试剂
硫酸、硝酸、高氯酸、硫脲(分析纯) 、硼氢化钾,、超纯水
仪器设备
微波消解仪、原子荧光光度计
1. 2 方法
湿消解(A)
称取标准物质小麦粉精确到0.01 g ,5 份,按GB/T 5009.11 -2003《食品中总砷及无机砷的测定》中氢化物原子荧光法前处理。
湿消解(B)
称取标准物质小麦粉精确到0.01g ,5 份,按GB/T 5009.11 -2003 氢化物原子荧光法前处理,但硝酸沸点121 ℃,高氯酸沸点203 ℃,硫酸沸点338 ℃,严格控制电热板温度在338~340 ℃之间,直到所有白烟冒尽,冷却后各加水25 ml ,继续蒸发至近干,但应避免完全干。
微波消解(A)
称取标准物质小麦粉精确到0.001 g ,5 份于微波溶样罐中,各加HNO3 5.0 ml 进行微波消解后,取出溶样罐,置于孔径于溶样罐相符的智能温控消解仪中,按HNO3 的沸点121 ℃进行赶酸至消解液形成像黄豆大小的液珠并可滚动为止。
微波消解(B)
同样按微波消解(A) 进行试样前处理,但赶酸至完全干枯。
2 结果与讨论
试样经4 种不同赶酸法前处理后, 按GB/T5009.11 -2003[1 ] ,氢化物原子荧光度法检测,结果见表1。
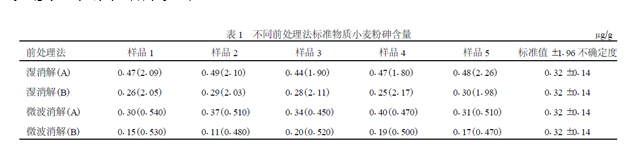
湿消解(A) 是完全按GBT5009.11 -2003 进行的,结果高于标准值,原因可能是没掌握好H2SO4 的赶酸沸点,消解液中还能嗅出刺鼻的酸味,说明此法赶酸是不彻底的,因此导致测定结果偏高。
湿消解(B) 是基于湿消解(A) 法,严格按H2SO4 的沸点赶酸,直至白烟冒尽,不易嗅出刺鼻的酸味,所以湿消解(B) 结果与标准值对照较接近(湿消解(B) 与湿消解(A) 经均数比较的t 检验(双测) , t = 15.53 v = 8 P < 0.001) 。但湿消解(B) 与微波消解(A) 相比,结果略低,这可能是由于敞开式湿消解易导致检测成分的蒸发所致。
微波消解(B) 结果很低,可能是由于赶酸至干枯所致。因为荧光光度法检测需在25 ml 内添加硫脲, 所以洗涤用水只能控制在约20 ml ,而20ml 水很难彻底把干枯在溶样罐壁上消解物彻底洗净。再者,蒸发至干枯也易导致被测成分的过量蒸发。
本次试验性研究微波消解(A) 法条件控制最佳,所以检测结果与标准值最接近(详见表1) 。由此可见,严格按照消解采用的酸的沸点控制赶酸温度,标准物质检测值在标准值可信范围。在原子荧光光度法检测过程中,常出现个别比色管污染导致本底残留对荧光光度法痕量分析污染残留权重放大的问题,由于荧光光度法软件有取样量与测定液定容体积比问题,尤其是在采用微波法处理试样时,取样量仅0.500 g ,消化液定容25 ml ,软件参数比为0.500/25 , 即: 1/50 。若本底残留为0.01μg ,则经软件换算处理就是0.50μg ,这样的结果在样本分析中所占权重是非常可观的。综上所述,试样的前处理是否规范将直接影响测定结果的准确性。
参考文献
[1 ] GB/T5009. 1~100 ―2003. 食品卫生检验方法理化部分[ S] .
最新消息
- 1赛多利斯新品Practum系列天平
查看次数:1283079......
- 22014年中国和世界十大科技进

由中国科学院、中国工程院主办,中国科学院院......
- 3天美中国与国家纳米科学中心

5月29日下午,天美(中国)科学仪器有限公司与......
- 4分析天平应该如何选砝码以及

查看次数:1107677 分析天平应该如何选砝......
- 5电子天平的使用与维护方法
电子天平是最新一代的天平,是根据电磁力平衡......