液相色谱法对牛、猪肌肉和肾脏中头孢噻呋残留量的测定研究
液相色谱法对牛、猪肌肉和肾脏中头孢噻呋残留量的测定研究
头孢噻呋(ceftiofur)是第一个动物专用的头孢菌素类抗生素,主要用于生畜呼吸道疾病的治疗。如果兽药不合理使用,会导致动物性食品药物残留超标,长期食用可引起细菌耐药性,造成人体微生物环境紊乱和失调。头孢噻呋在体内迅速代谢为脱呋喃甲酸头孢噻呋(desfuroylecefliofur,DFC)和呋喃甲酸(furoic acid),其中DFC为标志性残留物 。目前,欧盟和加拿大等一些发达国家规定头孢噻呋及其代谢物在动物组织中的最大残留限量(MRL):肌肉1 000 g/kg、肾6 000 g/kg。
头孢噻呋残留量的检测方法有微生物检测法、免疫分析法和液相色谱法 。微生物法样品处理简单、回收率较高,但不易筛选到特别敏感的菌株,缺乏特异性,而且多数分析周期较长。免疫分析法灵敏度高,特异性比微生物法强,适合于大量样品的快速筛选。液相色谱法(HPLC)是已报道用于头孢噻呋残留检测最常用的检测方法 。液相色谱一质谱法(LC―MS)可用于残留确证分析 ,但所用仪器要求高、价格昂贵。本文采用HPLC/UV法测定牛、猪肌肉和肾脏中的头孢噻呋相关残留量。与Jacobson等不同,采用多个固相萃取柱净化样品,虽然增加了操作步骤,但大大消除了样品基体对被测物的影响,检出限更低,且方法的回收率和精密度均较好。
1 实验部分
1.1 仪器与试剂
海能LC7000高效液相色谱系统,LC7011泵体,LC7020紫外一可见光检测器,LC7050自动进样器,配HanonClarity色谱软件。AutoSPE固相萃取装置。C18 固相萃取柱(500 mg,Va6an公司),使用前用4 mL甲醇和5 mL 0.025 mol/L磷酸缓冲液淋洗活化。SAX固相萃取柱(500 mg,Vafian公司),使用前依次用2 mL甲醇、2 mL甲醇一0.1 mol/L氯化钠(体积比25:75)和2 mL水淋洗活化。SCX固相萃取柱(100 mg,Varian公司),使用前依次用1 mL甲醇、2 mL甲醇一0.1 moL/L氯化钙(体积比25:75)和4 mL水淋洗活化。头孢噻呋标准品(纯度96.0% ,Dr.Ehrenstorfer公司);二硫赤藓糖醇(纯度99% ,Sigma公司);碘乙酰胺(纯度97% ,Sigma公司);三氟乙酸(纯度97%);乙腈、甲醇(色谱纯);其他试剂为分析纯,水由Milli Q净化系统(Millipore公司)制得。
1.2 溶液配制
头孢噻呋储备液(100 mg/L):称取10.0 mg头孢噻呋标准品于100 mL容量瓶中,用甲醇溶解并定容,一20℃保存,可使用6个月。头孢噻呋中间液(5 mg/L):吸取250 L 100 mg/L头孢噻呋储备液至5 mL容量瓶中,用0.025 mol/L磷酸缓冲液(pH 7.0)定容,摇匀(现配现用)。0.025 mol/L磷酸缓冲液(pH 7.0):将3.4 g磷酸二氢钾溶于900 mL水中,用450 g/L氢氧化钾溶液调节pH值至7.0,用水定容至1 L。0.05 mol/L硼酸缓冲液:称取19 g硼酸钠和3.7 g氯化钾,用水定容到1 L;4 g/L二硫赤藓糖醇溶液:称取1.0 g二硫赤藓糖醇,溶于250 mL 0.05 mol/L硼酸缓冲液中(现配现用);40 g/L碘乙酰胺溶液:称取2.0 g碘乙酰胺,溶于50 mL 0.025 mol/L磷酸缓冲液中(现配现用)。
1.3 色谱条件
C 柱(250 mm×4.6 mm,5 I.Lm);流动相:乙腈一水一三氟乙酸(体积比15:85:0.1);流速:1.0 mL/min;柱温:30℃ ;紫外检测波长:266 nm;进样量:25 L。
1.4 样品处理
1.4.1 提取与衍生称取1.0 g组织样品于50 mL离心管中,加入14 mL 4 g/L二硫赤藓糖醇溶液,均质,然后放入恒温振荡水槽仪中37℃保温20 min。加入3 mL 4%碘乙酰胺溶液,摇匀,室温下静置衍生30 min,离心10 min(4℃,20 000 r/min),取出上清液于25 mL比色管中。残渣中加入6 mL0.025 mol/L磷酸缓冲液,混匀,离心10 min(4℃,20 000 r/min),合并上清液,用25%磷酸溶液调节pH值至2.5~2.6,用水定容至25 mL。取出10.0 mL定容液,离心15 min(4℃ ,20 000 r/min)。
1.4.2 净化将“1.4.1”最后所得的上清液过C 固相萃取柱,保持流速1~2 mL/min,再依次用5mL磷酸缓冲液和3 mL 0.01 moL/L氢氧化钠溶液淋洗,用2 mL乙腈一水(体积比15:85)洗脱并收集于含5 mL水的试管中,保持抽真空2 min。取出收集的试管,加入5 mL水,摇匀,过SAX固相萃取柱,保持流速1~2 mL/min,用1 mL水润洗试管,加入柱中,用2.5 mL乙腈一体积分数为5% 乙酸(体积比5:95)洗脱并收集于另一含5 mL水的试管中,保持抽真空2 min。取出收集的试管,加入5mL水,摇匀,过SCX固相萃取柱,保持流速1~2 mL/min,用1 mL水润洗试管,加入柱中,用2.5mL乙腈一0.1 mol/L氯化钠(体积比5:95)洗脱并收集,保持抽真空2 min。取出收集的试管,摇匀,供测定。
1.4.3 标准品处理移取适量头孢噻呋中间液至50 mL离心管,加入14 mL 4 g/L二硫赤藓糖醇溶液,然后放入恒温振荡水槽仪中37℃保温20 min。加入3 mL 40 g/L碘乙酰胺溶液,摇匀,室温下静置衍生30 min,加入6 mL 0.025 mol/L磷酸缓冲液,用25%(体积分数)磷酸调节pH值至2.5~2.6,用水定容样液至25 mL。其余操作步骤同“1.4.2”节。
2 结果与讨论
2.1 衍生化
头孢噻呋在动物体内主要以脱呋喃甲酸头孢噻呋(DFC)代谢物存在,DFC可与体内生物大分子(如血浆蛋白)结合。结合态的DFC不容易被水性溶液提取,因此要将结合态变成游离态。二硫赤藓糖醇可以把蛋白结合的DFC及母体药转化为DFC,游离的DFC与碘乙酰胺反应,生成具有紫外吸收的衍生物脱呋喃甲酸头孢噻呋乙酰胺(DCA)。
2.2 净化条件的选择
提取、衍生后的样液净化采用C 柱、SAX阴离子交换柱和SCX阳离子交换柱3种不同的固相萃
取柱。C 柱主要去除中性杂质,SAX柱主要去除阳离子化合物,SCX柱主要去除阴离子化合物。本实验考察了1根柱(C 柱)、2根柱(C 柱和SAX柱)和3根柱(C 柱、SAX柱和SCX柱)的净化效果,发
现单独使用C 柱和使用C 与SAX柱的净化效果均不理想,样液中其它组分对测定有干扰。同时使用3根固相萃取柱,净化和富集效果较好(见图1)。
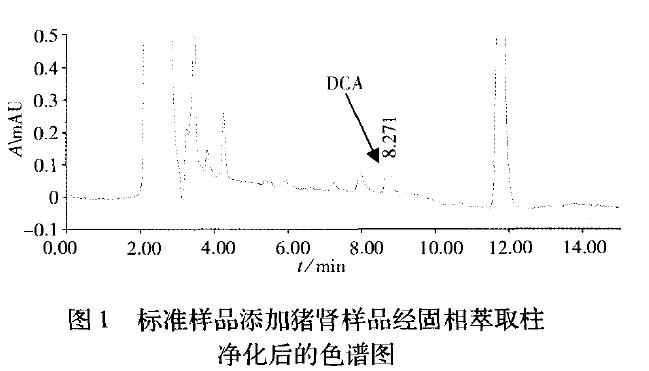
固相萃取柱的上样量对柱子的提取和净化有影响。实验发现,如果样液(25 mL)全部过柱,高浓度添加样品的回收率偏低,表明柱容量过载,有部分待测物流失。分取部分样液(10 mL)过柱,回收率明显提高,即使在高浓度时,也能满足定量检测的要求。
2.3 流动相的选择
流动相采用乙腈一水(该体系均含0.1%TFA)或甲醇一水(含0.1% TFA)体系,发现乙腈一水体
系优于甲醇一水体系。以乙腈一水(体积比10:90)和乙腈一水(体积比90:10)进行梯度洗脱,或以乙腈一水(体积比15:85)等度洗脱,头孢噻呋衍生物(DCA)的分离效果都比较好,样液中的其他组分对测定无干扰。由于梯度洗脱进样前需要平衡,分析时间较长,故选择等度洗脱。流动相中乙腈比例增加,头孢噻呋衍生物(DCA)出峰提前,但分离度变差。
2.4 头孢噻呋衍生物的稳定性
对1.28 mg/L头孢噻呋标准品衍生物(DCA)作稳定性试验,发现衍生物在4℃、避光条件下保存1个月,峰面积与刚衍生完时的峰面积几乎一致。
2.5 线性范围与检出限
在质量浓度0.016~1.28 mg/L(相当于添加量为100―8 000 kg)范围内,峰面积(y)与质量浓度( x)呈良好的线性关系,其线性方程为Y=5.016 7 +139.3,相关系数(r)大于0.999 9。根据3倍信噪比确定检出限为50 ug/kg。
本实验以4种动物性产品(牛肾、牛肉、猪肾、猪肉)作为样本。取适量标准品加入到1.0 g样品中,使添加量在牛肾和猪肾中相当于200、6 000和8 000ug/kg,在牛肉和猪肉中相当于200、1 000和2 000ug/kg,每个添加水平各取24份试样,按“1.4.1~1.4.2”节所述步骤进行处理,加标平均回收率为90%~91% ,相对标准偏差(RSD)1.8%~3.5% ,结果满意。
本方法经8家实验室的协同试验,肾脏样品添加量在200、6 000和8 000ug/kg和肌肉样品添加量在200、1 000和2 000ug/kg时(每个添加量单独测定2次),测得实验室间平均回收率为88%~91% ,相对标准偏差为4.2%~6.1% 。
2.7 应 用
取阳性样品牛肾和牛肉按“1.4”步骤进行处理和测定,由结果知,样品中其它组分对测定无干扰,据处理功能(间隔为0.5 nm)和Origin 7.5求积分功能,得到J=5.2×10-15 cm3・L ・mol-1;再由(2)式求出 值。一般认为药物对BSA的荧光猝灭作用主要源于212位色氨酸残基。当c(E):c(BSA)=1:1(相对分子质量以2 000计)时,E=1一, / =0.13,由(1)式求出R0值,得r=2.15 nm。
最新消息
- 1赛多利斯新品Practum系列天平
查看次数:1283079......
- 22014年中国和世界十大科技进

由中国科学院、中国工程院主办,中国科学院院......
- 3天美中国与国家纳米科学中心

5月29日下午,天美(中国)科学仪器有限公司与......
- 4分析天平应该如何选砝码以及

查看次数:1107677 分析天平应该如何选砝......
- 5电子天平的使用与维护方法
电子天平是最新一代的天平,是根据电磁力平衡......