ʳƷ����άƬ��������12 ��ά���صĸ�ЧҺ��ɫ��ͬʱ�ⶨ
ʳƷ����άƬ��������12 ��ά���صĸ�ЧҺ��ɫ��ͬʱ�ⶨ
����ά������һ�����ʸ���ĵͷ�������Ȼ�л������ ���в�ͬ���������ܡ����ӽṹ���������ʣ� ���������������о���ʮ����Ҫ�����ã� ����������������ͽ����������;ά���ص�����ܶ࣬ �����ܽ��Կɷ�Ϊˮ����ά���غ�֬����ά���ء�Ŀǰά���صIJⶨ������Ҫ�������ȷ�[1]��ӫ���ȷ�[2]������ɫ��[3]����ЧҺ��ɫ��(HPLC)[4��7]��Чëϸ�ܵ�Ӿ��[8��9]�ȡ�HPLC ����������㣬 �����Ժã� �������º͵IJ������������ȶ��Բ��ά����Ҳ�ɵõ�ȷ�Ķ������; ���ڶ���Ӧ��HPLC ����ά���ؽ��вⶨ��������ֻ�Ƕ�����һ��ά�����еļ��ֽ��вⶨ�� ��ˮ���ԡ�֬����ά���ؽ���ͬʱ�ⶨ�����ױ������١����о������˷����ЧҺ��ɫ��ͬʱ�ⶨ��8 ��ˮ����ά���غ�4 ��֬����ά���صķ����� ��Ӧ����ʵ����Ʒ�вⶨ�� ȡ���˽�Ϊ����Ľ����
����1 �����뷽��
����1.1 ��Ҫ�������Լ�
��������LC7000��ЧҺ��ɫ����; ɫ����(C18�� 5 ��m�� 4.6 ��250 mm) ;���״�(ɫ�״�)������(�ż���)���Ҵ�(������)����ˮ����(������)����������(������)����������(������)�������Լ���ָ�����Ϊ��������ά���ر�Ʒ�� VC��VB1��VB2��VB6��VB12�����ᡢҶ�ᡢVD3��VE ��Ʒ; ��������Ʒ; VK1 ��Ʒ��; VA (�ӻƴ�) ��Ʒ��
����ά���ر�����Һ�� �ֱ��ȡά����A��D3��E��K1�ı�����Һ(1 mg / ml�� �Ҵ�����)�� -18��ܹⱣ��; VB1��VB2��VB6��VB12�����ᡢ������������Һ(1 mg / ml�� 0.01 mol / LHCl ����); Ҷ�������Һ(1 mg / ml�� 200 ��l 5 mol / L KOH�ܽ�� ����0.01 mol / L HCl ����); VC ��������(1 mg / ml��0.01 mol / L HCl ����); ����Һ������-18������С���������ά���ر�Ӧ��Һ��
����ʵ����ˮΪMillipore ����ˮ(18.2 M����cm)��֬����ά����A ��E Ӧ��ҺŨ���ڲⶨǰ�궨[10]��
������ʵ���еĶ�άƬ��ǿ���ۡ����ۺ�������Ʒ�����Գ��С�
����1.2 ʵ�鷽��
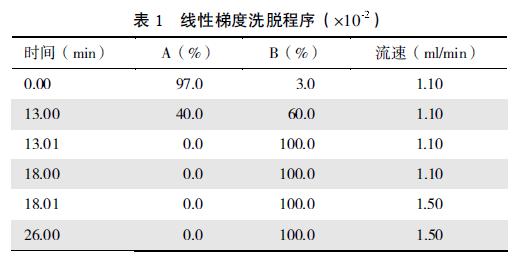
����1.2.1 ɫ�����������ࣺ A�� 0.05 mol / L KH2PO4��Һ(pH 6.0); B�� �״�; ɫ�������ݶ�ϴ�ѳ���; ���£� 40�档����1��
����1.2.2 �����ߵĻ��Ƹ��ݲ�ͬ��Ʒ��ά���صĺ�����Χ���ñ�����Һ���Ʋ�ͬŨ�ȵĻ�ϱ�Һ�� �ڸ�ЧҺ��ɫ���ǻ��ߴ�ƽֱ�� ���вⶨ�����Ʊ�����; ά���ػ�ϱ�ɫ��ͼ��ͼ1��
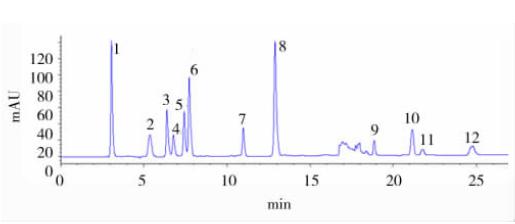
����ע�� 1. ά����C (VC�� 3.0 min); 2. ����(Niacin�� 5.3 min); 3.ά����B1 (VB1�� 6.3 min); 4. ά����B6 (VB6�� 6.7 min); 5. ������(Nicotinamide�� 7.4 min); 6. Ҷ��(Folic acid�� 7.8 min); 7. ά����B12(VB12�� 11.0 min); 8. ά����B2 (VB2�� 12.9 min); 9. ά����A (VA��18.7 min); 10. ά����D3 (VD3�� 21.1 min); 11. ά����E (VE�� 21.7min); 12. ά����K1 (VK1�� 24.9 min)��
����ͼ1 12 ��ά���ػ�ϱ���Ʒɫ��ͼ
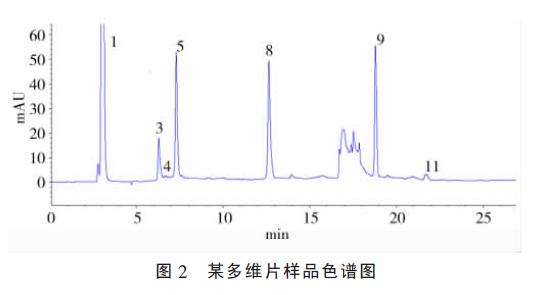
����1.2.3 ��Ʒ��������ά����Ƭ������ȷ��ȡ0.2 g ��ĥ���ȵ���Ʒ��15 ml ���Ĺ��ڣ� ����10 ml 0.01 mol / L HCl��������ȡ15 min ��12 000 r / min ����5 min�� ȡ����Һ��Ϊ����ҺA; ��ȷ��ȡ��Ʒ(��ĥ���ȵĸ���ά����Ƭ0.7 g ����4 g) ��250 ml ����ƿ�У� ����20 ml �Ҵ��� ������ȡ15min�� ����50%��KOH ��Һ(����ά����Ƭ��5 ml; ��10ml)�� 90������30 min�� ����Һ��100 ml ������ȡ����40��ˮԡ�������ɣ� 5 ml �Ҵ����ݣ� ��Ϊ����ҺB; ��ͬ����Ʒ��AҺ��B Һ�������9�U1 �����Ϊ����Һ�� 20 ��l ��������; ��Ʒɫ��ͼ��ͼ2��3��
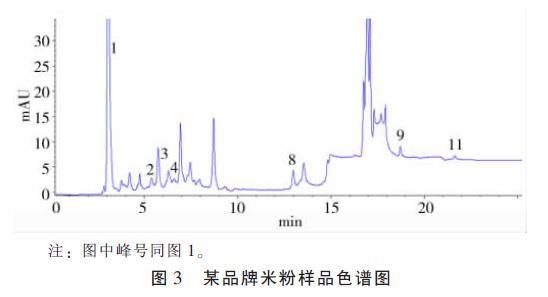
��������ȡһ������0.45 ��m ��Ĥ���˺� 20 ��l ������������Ʒɫ��ͼ��ͼ4��
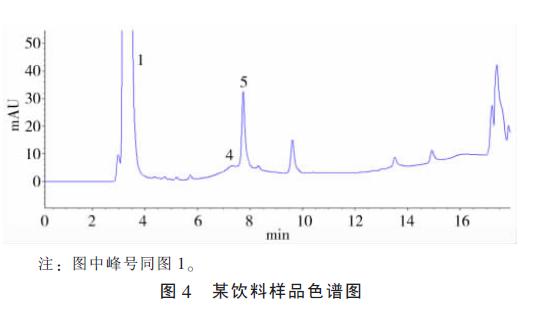
����1.2.4 ��Ʒ�ⶨ���������ڱ���Һ�ⶨ��ͬ�����£� ����Һ���вⶨ���Ա���ʱ�䶨�ԣ� ��ȡ��ά���ط������ͨ���ع鷽�̡���Ʒ����������ȡҺ���������Ʒ�и���ά���صĺ���(mg / kg)��
����2 ���������
����2.1 ɫ���������Ż�
����2.1.1 �ݶ�ϴ���������Ż�������[11]�Ļ����ϣ� ѡ��״����������ػ���ҺΪ������; ��ȶ�ϴ�ѷ������ѣ� �����ݶ�ϴ�ѷ�ʽ������12 ��ά����ɫ���Ρ�����Ⱥ�ɫ����ʱ�䣬 ����ȷ�������ܼ���ɱ������ݶ�ϴ�ѳ������1��
����2.1.2 �������л���ҺpH ֵ��ѡ������ά���ص����ʲ��ϴ� ������pH ֵ����ɫ��������ҪӰ��; ����ҺpH ֵΪ4.0��8.0 ʱ�� ������2 ��ά���س���ʱ�䷢���غ�; ��pHΪ6.0 ʱ�� 12 ��ά�����ܹ���ȫ���롣
����2.1.3 ���µ�ѡ��ʵ������� ������Ϊ30�桫40��ʱ�� ��ά���ط������� ������30��ʱ�� VB1��VB6����������Ҷ�����ȫ���룬 ���з�չ������; ����Ϊ40��ʱ�� VB1��VB6����������Ҷ��ﵽ��ȫ�����ҷ��νϺ�; �¶�50��ʱ��VC ��������Ա�С�� ��VB1��VB6����������Ҷ����غ�;��ˣ� ѡ��40��Ϊɫ���������¶ȡ�
����2.1.4 ���ٵ�ѡ��������̶��� ���ٵ�ѡ���Ӱ��ɫ���룬 ����ˮ���ԡ�֬����ά�������ʲ�ͬ�� ����ڷ���������ѡ���˲�ͬ�����ټ���1��
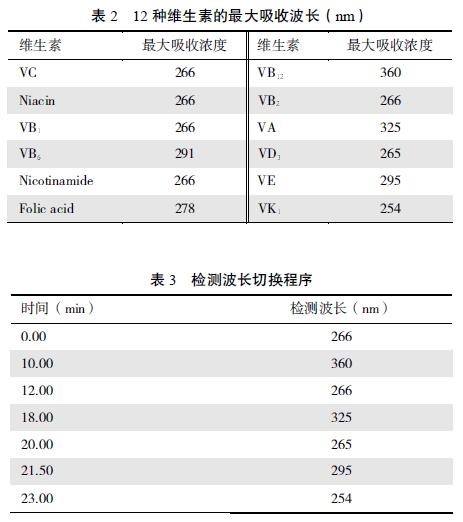
����2.1.5 ��Ⲩ����ѡ��8 ��ˮ����ά���ص�������ղ������(����2)�� ����VB1��VB6��������Ҷ��ķ����ʱ�����϶̣� ������ɲ����л��� Ϊȷ�����������ȣ� ��VB1��VB6����������Ҷ��ļ�Ⲩ������Ϊ266 nm; �����Ϊ��������ղ�������Ⲩ���л��������3��
����2.2 �ⶨҺ���Ҵ������Բⶨ��Ӱ��
������ά���ػ�ϱ�Һ�У� �Ҵ�������VA ��������������ã� ����11 ��ά���ط����������Ӱ�졣�����Ҵ���������
����10%ʱ�� ������ο�ʼ�ֲ棬 �����Ҵ���������ߣ� VB1��VB6�ķ��κͷ����Ҳ�ܵ�Ӱ��; ������̫�� ���ᡢVB1��VB6����������Ҷ��ķ��ζ�Ҳ�б������ơ����ڲⶨҺ����̶��Ҵ�������һ���ķ�Χ��
����2.3 ������������
����2.3.1 ���Է�Χ�ͼ�������Ż���ɫ�������£� ���������Իع鷽�̡���ͼ���ޡ�����ʱ��ͷ��������Ա������4��

����2.3.2 ���ܶȱ�����������ռ侫�ܶȷ�ΧΪ1.35%��4.11%����Ʒ�ⶨ���ܶȲⶨRSD ��ΧΪ0.92%��4.80%��
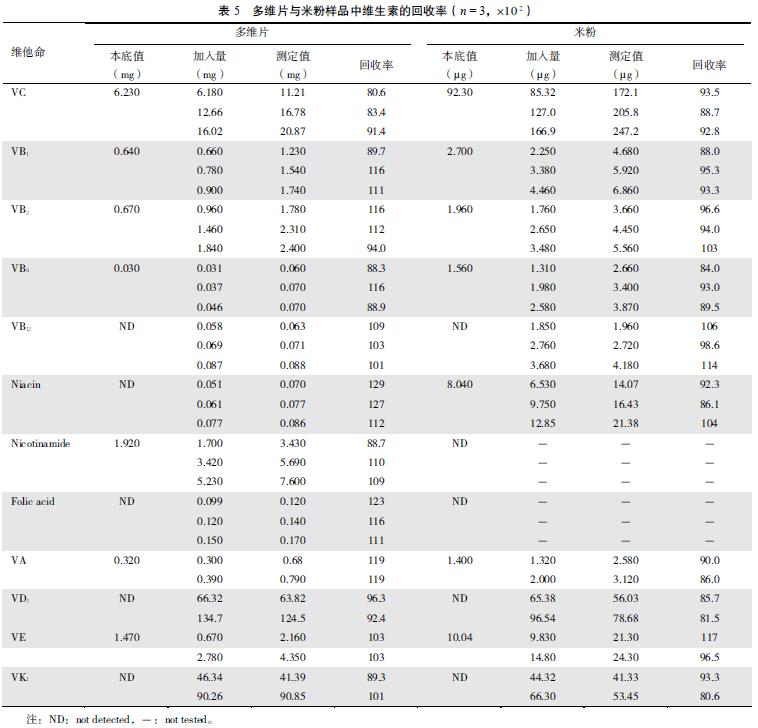
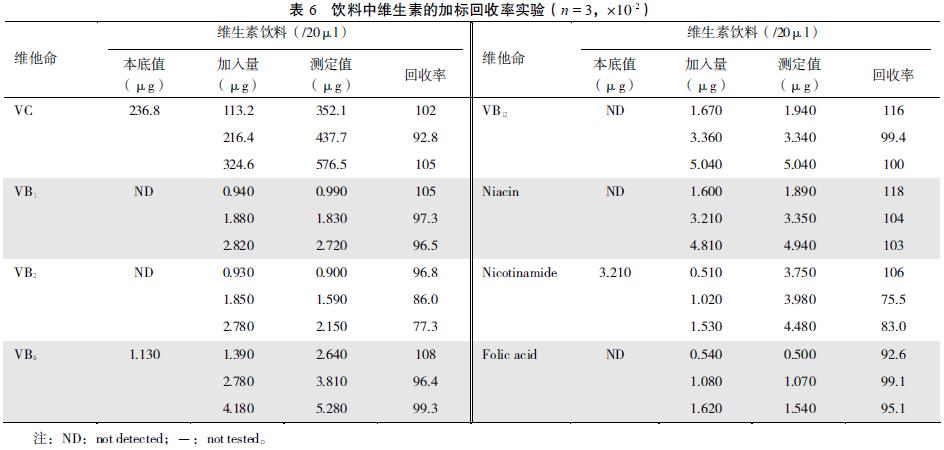
����2.3.3 ȷ���ɱ�5��6 �ɼ��� �����ڶ�άƬ���ۺ�������Ʒ�и�ά���صļӱ�����ʷ�Χ��75.5%��129%��
����2.4 ����Ӧ��
����Ӧ�ñ����ⶨ�˸���ά����Ƭ���ۡ����ϡ�������ˮ���Ժ�֬����ά���غ����� �������7��8��
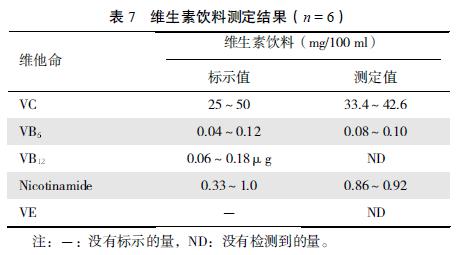

�������ϣ� ���ı�����ʳƷ�������ж���ά���صķ����ЧҺ��ɫ��ͬʱ�ⶨ������ ʵ����һ�δ�����ͬʱ����12 �ֲ�ͬ����ά���ص�Ŀ�ģ� ����һ����ʵ��Ӧ�ü�ֵ��
������Ϣ
- 1������˹��ƷPractumϵ����ƽ
�鿴����:1283079......
- 22014���й�������ʮ��Ƽ���

���й���ѧԺ���й�����Ժ���죬�й���ѧԺԺ......
- 3�����й����������ѧ����

5��29�����磬����(�й�)��ѧ��������˾��......
- 4������ƽӦ�����ѡ�����Լ�

�鿴����:1107677 ������ƽӦ�����ѡ��......
- 5������ƽ��ʹ����ά������
������ƽ������һ������ƽ���Ǹ��ݵ����ƽ��......