高效液相色谱法测定化学合成生物多肽
高效液相色谱法测定化学合成生物多肽
多肽类化合物广泛存在于自然界中,其中对具有一定生物活性的多肽的研究,一直是药物开发的一个主要方向。80年代中期至今多肽化学合成技术有很大发展,合成的成品一般是以目标多肽为主的几种结构相似多肽的混合物,对多肽混合物的分离分析及纯度鉴定一直是一个重要问题。本文主要介绍本人在分析多肽类化合物过程中的一些心得和经验.
首先,在色谱柱选择方面,因为多肽的分子量较大(一般在几千到几万),分子量在10000左右的,常用的C18柱分离效果不太好,要选用碳链比较短的色谱柱,如:C8或者C4;填料孔径不能选普通的100埃,要选孔径比较大的如300埃等。分子量在几万或者几十万的,可以选体积排阻色谱,用凝胶色谱柱。
其次,在用反相色谱柱时,因为多肽是由氨基酸组成的,流动相选择一般用含0.1%的三氟乙酸水和含0.1%的三氟乙酸乙腈的梯度洗脱。但因为多肽类化合物的紫外吸收较弱,检测波长一般选在200~210nm左右,所以空白梯度的基线会往上漂移很多,特别是样品浓度比较小的时候,分析误差较大。考虑乙腈的本底吸收比水大,在梯度洗脱过程中,随着乙腈比例的增大,基线会向上漂。因此把乙腈中的三氟乙酸浓度降为0.05%~0.07%(可以随梯度不同选不同浓度),通过两者紫外吸收的抵消,基线变得平稳很多,同时能增加检测的灵敏度,使分析更灵敏、更准确。
第三,合成的多肽中的杂质一般与产物结构相似,极性相差无几,分离难度较大,在选择洗脱梯度时要细心一点,多做一些试验,最好要做主峰的纯度扫描,尽量把杂质完全分开,对于难分离的多肽,可以选长一点的色谱柱或者延长梯度洗脱的时间。
还有最后一招,如果按以上方法分离还是不太好的话,可以适当的提高柱温,我们有一品种就是这样的,当柱温升到60度时,塔板数和与杂质的分离度都能得到一定的改善。
HPLC 分析柱有两类:离子交换柱和反相柱。离子交换 HPLC 依靠多肽和固相间的直接电荷相互作用,柱子在一定 PH 范围带有特定电荷衍变成一种离子体,而多肽或多肽混合物,由其氨基酸组成表现出相反电荷,分离是一种电荷相互作用,通过改变 PH 值、 离子强度,达到分离纯化的效果。通常,先用低离子强度的溶液,以后逐渐加强或一步一步加强,直到多肽从柱中洗脱出。
反相 HPLC 条件与正常层析正相反,多肽通过疏水作用连到柱上,用降低离子强度洗脱, 如增加洗脱剂的疏水性。通常柱子由共价吸附到硅上的碳氢烷链构成,这种链长度为 G4-G8 碳原子。 由于洗脱是一种疏水作用。长链柱比短链对小的,高带电肽好;另一方面大的疏水肽用短链柱洗脱好。
典型的操作常由两绶冲剂组成, 0.1%TFA-H2O 和 80% acetonitrile 0.1%TFA--H2O 稀 acetonitrile 。用线型梯变以每分钟 0.5% 到 1.0% 改变的速度混合。常见分析和纯化用柱为 4.6× 250mm (3-10μ m) 和 22× 250mm (10μ m). 如果用径向填柱,那么大小是 8×100 ( 3-10μ m )和 25× 250mm (10μ m)
大量各种缓冲剂含许多不同试剂,比如 heptafluorobutyric 酸, 0.1% 磷酸, 稀 Heformic 酸 (5-6%, pH2-4), 10 -100mM NH4HCO3, 醋酸钠 / 氨, TFA/TEA ,磷酸钠或钾,异戊酚。这样许多不同组合可形成缓冲剂,但要注意一点:硅反相柱料不能长时间暴露于高 pH ,甚至微碱 pH , 因为这样会破坏柱子
一般多肽分离拿150mm的柱子就可以了,250mm在分离度上改善不大,因为多肽的分离机理和小分子不同。
小分子是通过液液交换,所以柱子越长交换越充分,分离度也越高。
多肽在柱子上的保留和分离其实主要发生在柱头,也就是说,多肽先在柱头都被保留下来,随着梯度上升逐渐被洗脱,所以柱长对多肽分离的影响比起其对小分子的分离度影响小得多。
最新消息
- 1赛多利斯新品Practum系列天平
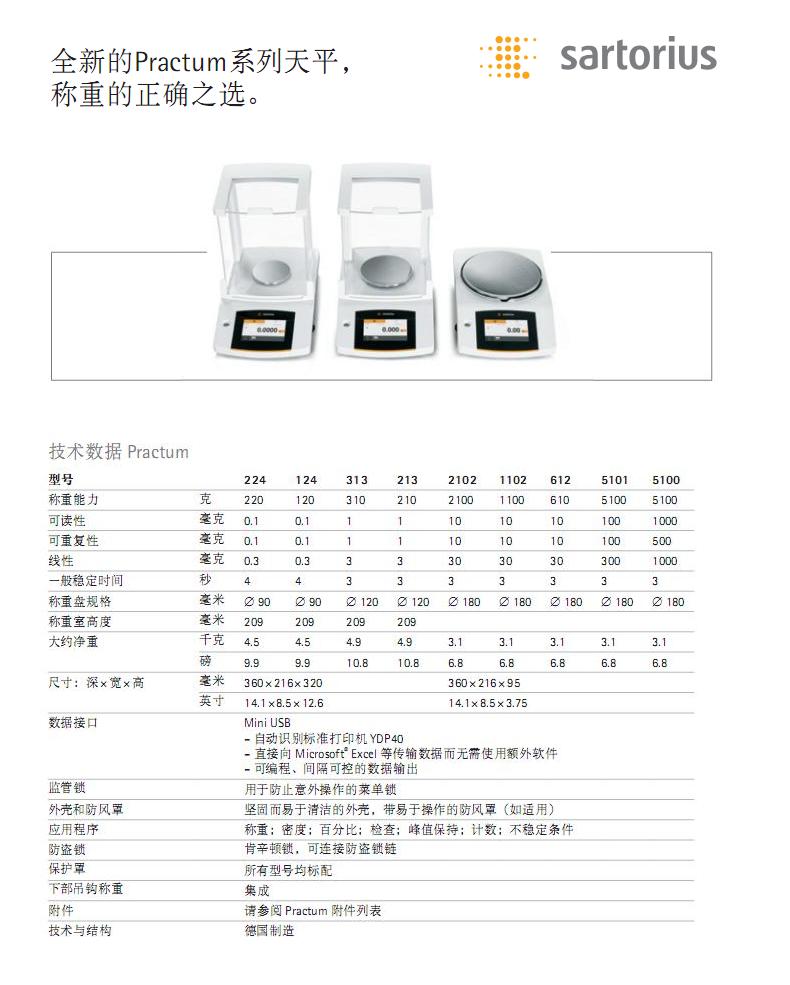
查看次数:1283079......
- 22014年中国和世界十大科技进

由中国科学院、中国工程院主办,中国科学院院......
- 3天美中国与国家纳米科学中心

5月29日下午,天美(中国)科学仪器有限公司与......
- 4分析天平应该如何选砝码以及

查看次数:1107677 分析天平应该如何选砝......
- 5电子天平的使用与维护方法
电子天平是最新一代的天平,是根据电磁力平衡......