���յ�����ȡ���ЧҺ��ɫ�����ü�����Ʒ�еĶ����
���յ�����ȡ���ЧҺ��ɫ�����ü�����Ʒ�еĶ����
ժ Ҫ �����Ľ�����һ���Ա��ͦ�-������ˮ��Һ��Ϊ��ȡ�ܼ��Ķ���Һ����ȡ-��ЧҺ��ɫ������������·�����ͨ����-�������������İ������ö���Ʒ�е����ְ�ӷ��Զ���������˷��븻������Ӱ����ȡЧ�ʵ���Ҫ���ؽ����˿��졣�����ŵ������£��÷��������ֶ����(�����ƣ��죬ӫ�����)�ļ���ֱ�Ϊ0.097, 0.016, 0.004, 0.247, 0.098ng/ml����Ա�ƫ��(RSDs)Ϊ5.1-7.1%( n=7)��ʵ������������-������������߶Զ��������ȡЧ�ʣ�������ǿ��ЧҺ��ɫ��ӫ�����źš�������Ӧ���ںϳ�ˮ���ͻ�����Ʒ(ˮ������Ҷ��Ʒ)�ķ���������������⡣�÷��������٣�ѡ���Ըߣ��������л��ܼ��������ڸ��ӻ����к���������ķ�����
�����ؼ��ʣ����յ�����ȡ;��ЧҺ��ɫ��;�����;��-������;������Ʒ
����1 ����
���������(Polycyclic aromatic hydrocarbons�����PAHs)�ǻ������ձ���ڵ�һ�ֳ־�����Ⱦ����������ǿ�ҵ��°��ͱ������ã�������������(���EPA)ȷ����16 ��PAHs��Ϊ���ȼ����Ⱦ�PAHs ��Ҫ��Դ��ʯ�͡�ú��ȼ�ϼ�ľ��,��ȼ����IJ���ȫȼ�ա�
�������ڻ����зֲ��ܹ㣬PAHs ͨ��������ˮ��ʳƷ�����̵Ƚ������壬���ص�Ӱ������Ľ�������ˣ����ǶԶ�������о��������㹻������[2,3,4]��ȷ�ⶨ�����ж����������ĺ�������ʮ����Ҫ�����壬Ȼ���ڻ����ж�������ڵ�Ũ�Ⱥܵͣ����һ�����Ʒ���帴�ӣ�������࣬������ֱ�Ӳⶨ��ͨ����Ҫ����Ʒ����ǰ��������ˣ�Ѱ�ҿ��١���㡢��Ч���������ȵ�ǰ��������һֱ������ѧ�о����ȵ㡣
����Ŀǰ�����ڲⶨ�����ж��������Ʒǰ���������кܶ࣬���б����ķ����У�������ȡ(Soxhlet extraction��Һ-Һ��ȡ(liquid-liquid extraction)����������ȡ(ultrasonicextraction)�������ܼ���ȡ(accelerated solvent extraction, ASE)����������ȡ(microwave assisted extraction, MAE)�����ٽ�������ȡ(supercritical fluid extraction, SFE)������ȡ(solid phase extraction, SPE)[9-12]��ǰ���ַ�������Ҫ���Ĵ�������
�������ܼ����Ի�����ɺܴ��ѹ�������ҷ�ʱ�������ַ���������Ч�����л��ܼ������������̲���ʱ�䣬����Щ������ʹ������������豸�������ɱ��ߡ�
����Һ����ȡ������һ�ֿ�ݡ���Ч���ͳɱ����ܼ��������ٵ���Ʒǰ�����������÷��ɷ�Ϊֱ��Һ����ȡ�Ͷ���Һ����ȡ[17]����ģʽ�����ж���Һ����ȡ�����ڷ�������������Ʒ�Ϸ��ռ�Ļӷ��Ի��ӷ����л��������ֱ��Һ����ȡ��ȣ�����Һ����ȡ��������Ʒ�����Ӱ�죬��ˣ��÷����㷺Ӧ���ڻ�����Ʒ�еĻӷ������ӷ����л���Ⱦ��ķ���[17-20]��
������������һ���������ǵ�Ԫ��ɵĴ���������ڲ���һ��Ѩ��������ɵĵ�Ԫ������ͬ����Ϊ�����£��� �������͡������������ⲿ������ˮ�ԣ�����ˮ�о���һ�����ܽ�ȣ������ڲ���Ѩ������ˮ�ԣ��������Ծ����������Ը��ݿ�����ӵĴ�Сѡ���Եؽ���л����ӣ��γɾ��в�ͬ�ȶ��̶ȵİ�����[21,22]��1988 �꣬Blyshak[23]�Ȳ��æ�-��������Һ��ȡ������еĶ���������p���F����ȡЧ�ʿɴﵽ90 %���ϡ����о������������������Ը������ǻ��Сѡ���Ե���ȡ�ض��Ķ������Husain ��[24]���о���������-�������ܹ���ǿ�����ӫ�����ӫ��ǿ�ȡ�
�������ĵ�Ŀ���ǽ���-������ˮ��Һ��Ϊ������ȡ����ȡ�ܼ���ͬʱ�ֽ�����Ϊӫ���ź���ǿ�Լ�����������Һ����ȡ�������ЧҺ��ɫ��ӫ���⼼��(HPLC-FLD)���ã��ⶨ������Ʒ�е����ֶ����(�����ƣ��죬ӫ�����)���·�������������ϸ̽���˶���Һ����ȡ��ϵ����Ҫʵ�����(�磺��Ʒ�¶ȣ���������������ٶȣ���ȡʱ�䣬����ǿ�ȵ�)����ȡЧ�ʵ�Ӱ��;�������������·������ڻ�����Ʒ�ж�����IJⶨ��
����2 ʵ�鲿��
����2.1 �Լ��ͱ���Һ
������(������)����(��ѧ��)�ֱ�������к��˻�ѧ�Լ���������˾������й⸴��ϸ�����о�����������ֽ������ṩ����(98%)��ӫ��(95%)����ɽ���쳤��������˾.
����������Һ�ܽ��ڼ״����Ʊ����ɣ������ƣ�ӫ�죬��(1 mg/mL);��(0.25 mg/mL)��
��������Һ��4 oC �����±��档
�����״�(������)����ɽ��������������ʹ��ǰ��0.45��m ��©�����ˣ���-������(������)����Sigma Aldirich ��˾��
����ʵ�������õ������Լ���Ϊ�ż������������18.2 MΏ �ߴ�ˮ(Labconco system)��������ʵ����̡�
��������������ʹ��ǰ����10%�����н���24 Сʱ���ϣ�ʹ��ǰ�øߴ�ˮ��ϴ�ɾ���
����2. 2 ��Ʒ����
������Ȼˮ����ȡ��������ˮ(pH 7.7���人)������ǰ����0.2 g/ml ���Ȼ��ƣ������������pH ֵ��5.0������ȡֱ��ע���ЧҺ��ɫ���з����⡣
������Ҷ���ɼ�ij��������������Ҷ�����顣��ȷ���ص���Ҷ��pH ֵΪ5.0 ����������/�������ƻ�����Һ(��0.2g/ml ���Ȼ���)����24 Сʱ���پ���ȡֱ��ע���ЧҺ��ɫ���з����⡣
����2. 3 ����Һ����ȡװ�ü���ȡ����
�������IJ��ö���Һ����ȡװ�õ�ʾ��ͼ��1 ��ʾ������40oC ��ˮԡ���ȣ���װ��10 mL��Ʒ��Һ�Ķ���ƿ���ں��´���������(�����ٻ�������������˾)�����Ϸ���ˮԡ�ձ����������������ȡ10��L �ı��ͦ�-��������Һ��Ȼ����Һ�ڶ���ƿ�Ϸ�������⣬��ȡһ��ʱ�����Һ�Σ�ֱ��ע���ЧҺ��ɫ���з����⡣
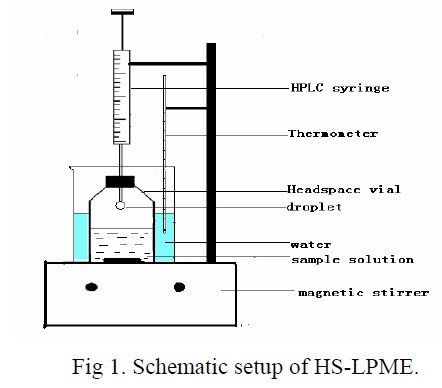
����2. 4 HPLC ϵͳ
������������ʹ�õĸ�ЧҺ��ɫ����ΪHanonLC7000��Ԫ��ѹɫ��ϵͳ��ɫ����ΪRP-C18 ��(ODS��250mm��4.6 mm ��5-��m ����)��ӫ����IJ������ڱ�1���Է�������������õȶ�ϴ�ѣ�������Ϊ90%�״���10%ˮ��������Ϊ1 mL/min �������£����ֶ�����ܵõ���Ч�ط������⡣
����3 ���������
����������Ӱ�춥��Һ����ȡЧ�ʵ����أ�������������������ʣ���ȡʱ�䣬��Ʒ�¶ȣ�����ǿ�ȣ��Լ�pH ֵ�ͦ�-������Ũ�ȡ�
����3.1 ����Һ����ȡ�����Ż�
����3.1.1 ��Ʒ�¶ȵ�Ӱ��
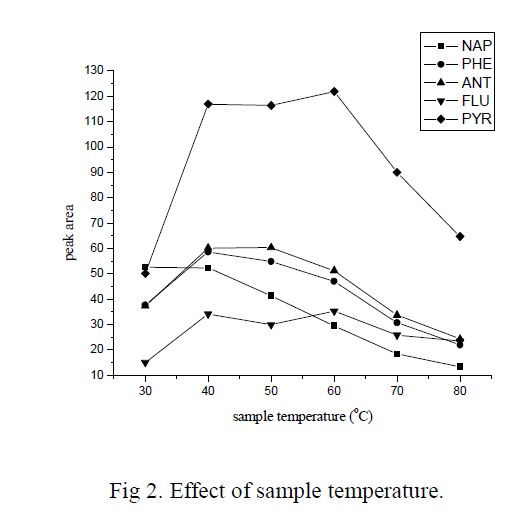
�������ڶ�����ȡ��ӷ���������˵���¶���һ����ҪӰ�����ء��ڱ�ʵ���У�ʹ�ú���ˮԡ������Ʒ��Һ���¶ȣ�������30oC-80oC ��Χ���¶ȶ���ȡЧ�ʵ�Ӱ�죬�����ͼ2 ��ʾ�������¶ȵ����ߣ����ֶ�������ź�ǿ�ȶ������Ե���ǿ�������¶ȴ���40oC ʱ��ӫ�죬�ŵ���ȡЧ�ʲ������������ƣ������ȡЧ�ʷ������͡�һ������£������¶ȿ��Լӿ�����˶�, ����Һ������ѹ������, ������������ȡ��ƽ��ʱ��[25]; ��һ����,��-�������������İ�����һ�����ȵĹ���[21]����ˣ������¶ȷ���ʹ����ȡЧ���½����ۺϿ��Ƕ���ѧ������ѧ��Ӱ�죬ʵ����ѡ��40oC ��Ϊ��Ʒ�¶ȡ�
����3.1.2 ���������Ӱ��
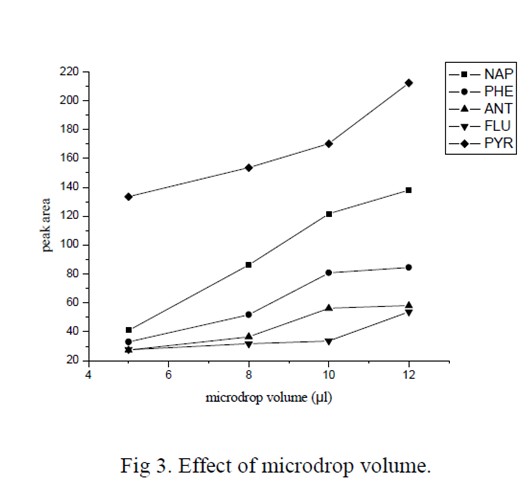
��������Һ����ȡһ������л��ܼ���Ϊ��ȡ�ܼ�����ʵ����û�����ˮ��Һ��Ϊ��ȡ�ܼ���ʵ������ȫ��ɫ����ȡ��������ȡ������Һ�μ���û�лӷ���ʧ��ʵ������������������ͷ������һ����Һǹǹͷ��ʹ����ͷ�Ͽ������ҽϴ������Һ�Ρ���ͼ3 ��ʾ����Һ�������2-12��L �ķ�Χ�ڱ仯ʱ����ȡЧ����������������������Ȼ������Һ���������10��L ʱ���������������������ã�Һ�α�ò��ȶ�����˱�ʵ����õ���ȡ�����Ϊ10��L��
����3.1.3 �������ʵ�Ӱ��
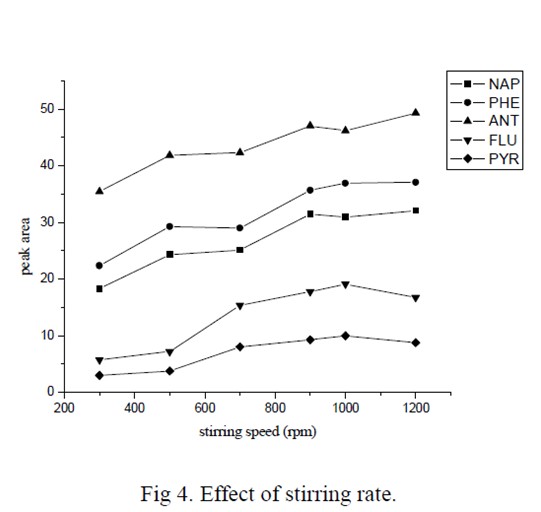
������߽������ʿ��Լ���ˮ���з�����Ļӷ�������̬��ʽ������Ʒ�ϲ��ռ䣬����ƽ��ʱ�䣬�����ȡЧ�ʡ���ͼ4 ���Կ��������Ž������ʵ����ӣ����������ȡЧ�������Ե�����ת�ٴ���900 rpm ʱ����ȡЧ�ʱ仯�����ԡ���ˣ���ʵ��ѡ��1000 rpm ��Ϊ��ȡ�������ʡ�
����3.1.4 ��ȡʱ���Ӱ��
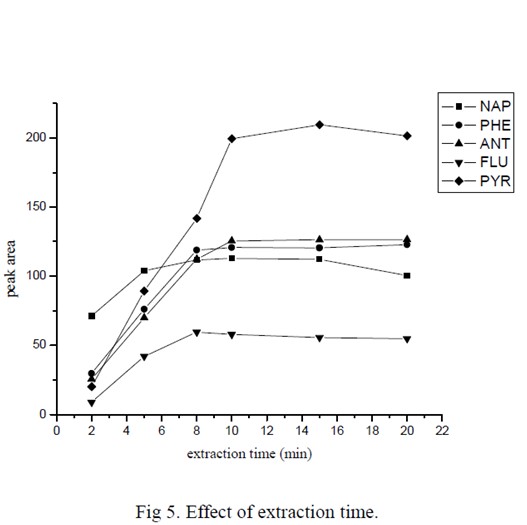
�������������û�������PAHs �İ�������ʵ����ȡ����ˣ���ȡЧ���ںܴ�̶����ɰ������õ�ƽ�ⳣ��������ͼ6 ����ȡʱ���5 ��PAHs ��ȡЧ�ʵ�Ӱ�졣���Կ������������ź�ǿ����10 min �Ժ�Ͳ�������˵����ʱ��ȡ�Ѿ������ϴﵽƽ�⡣���������ź�ǿ����5 min ��ͻ����������ȶ���������Ļӷ�����������Ǻϵġ���������ʵ����������ѡ�����ȡʱ��Ϊ10 min��
����3.1.5 ����ǿ�ȵ�Ӱ��
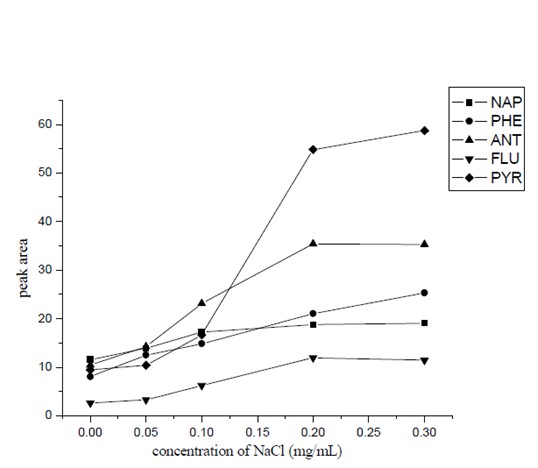
��������Ʒ��Һ�м���������Լ�С�л�����ܽ�ȣ��Ӷ�ʹ����Ĵ�������붥�������ȡЧ�ʡ��ڱ�ʵ���У������˼����Ȼ��Ƶ�������ȡЧ�ʵ�Ӱ�졣��ͼ6 ���Կ����������Ȼ��Ƶ�Ũ����0-0.2 g/ml ��Χ�ڱ仯ʱ��PAHs ����ȡЧ�������Ȼ���Ũ�ȵ����������������Ȼ���Ũ�ȴ���0.2 g/ml�������Ȼ���Ũ�����ӣ���ȡЧ�ʱ仯�����ԡ���ˣ��ڱ�ʵ���м����Ȼ��Ƶ�Ũ��Ϊ0.2 g/ml��
����3.1.6 pH ֵ�Լ���������Ӱ��
����������Һ��pH ֵ�ܹ��ı����������Һ�еĴ�����ʽ��ʵ��������������/�������ƻ�����Һ������Ʒ��pH ֵ����pH4-10 �ķ�Χ�ڣ��о���pH ��PAHs ����ȡЧ�ʵ�Ӱ�졣
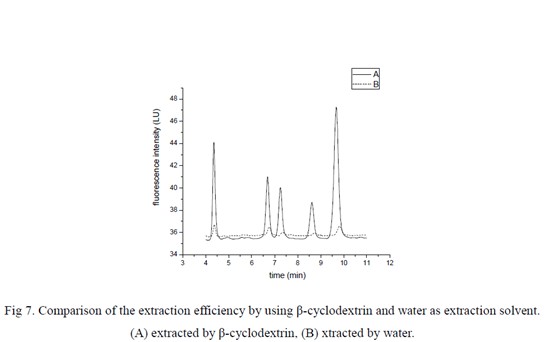
������������ڵ�pH ��Χ���нϺõ���ȡЧ�ʣ���ˣ�������ѡ��pH ֵ5 ��Ϊ��ʵ����Ʒ��Һ��pH ֵ����ʵ���о��˦�-����������ȡЧ�ʵ�Ӱ�졣ʵ���зֱ���8��l ��ˮ�ͦ�-����������ˮ��Һ������ȡ�������ͼ7 ��ʾ������A ��8��l �ı��ͦ�-������ˮ��Һ��ȡ����ȡ�����ټ���8��l ��ˮ����Ͼ��Ⱥ�ע��HPLC ������;����B ������8��l ��ˮ��ȡ����ȡ����ע��HPLC ֮ǰ����8��l �ı��ͦ�-������ˮ��Һ�����Կ�����A ����õ��ź�ǿ��ԶԶ����B����õ��ź�ǿ�ȣ����������-��������PAHs �кܺõ���ȡЧ����
����ͬʱ��ʵ�黹�о��˦�-������Ũ�ȶ���ȡЧ�ʵ�Ӱ�죬���ֶ��������ȡЧ�ʶ����Ŧ�-������Ũ�ȵ��������������ڱ�ʵ��������ѡ��ʹ�ñ��͵Ħ�-������ˮ��Һ��Ϊ��ȡ�ܼ���
����3.2 ��-��������ӫ���źŸ�������
����ʵ���п�������ͬŨ�ȵĶ�����ڲ��Ӻͼ���0.01 M��-������ʱ��ӫ���ź�ǿ�� ����ͼ8 ��ʾ���ڦ�-�����������µ�����£��������ӫ���ź�ǿ�Ⱦ������Ե���ǿ����-���������������Կ��Լ�����߷����������ȡ�
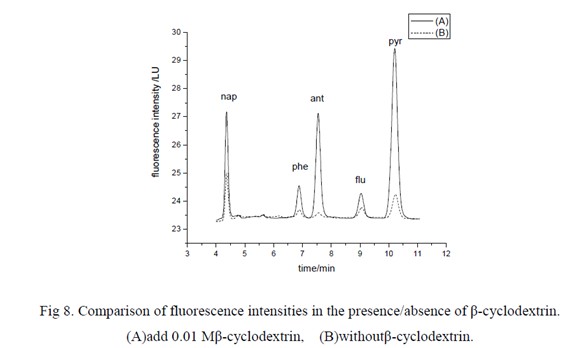
����3.3 ��������
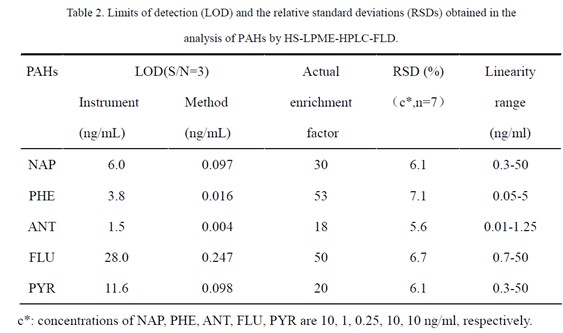
�����Է����ķ������ܽ����˿��죬�������2�������������Է�ΧΪ0.01-50 ng/ml ֮�䣬��Ӧ����Ա�ƫ��(RSD s)������10%;���������ֱ�Ϊ18-53 ��;�����(S/N=3)��0.004-0.247 ng/ml ��Χ�ڡ�
����3.4 ��Ʒ����
����3.4.1 �ϳ�ˮ����ʵ��ˮ��
�������������ķ������ںϳ�ˮ���ķ�������֤������ȷ�Ⱥ;��ܶȣ����ý�����ڱ�3�С����Կ�������PAHs �ļ���ֵ��0.05-10 ng/ml �ķ�Χ�ڣ��ⶨֵ������ֵ�ǺϽϺá�
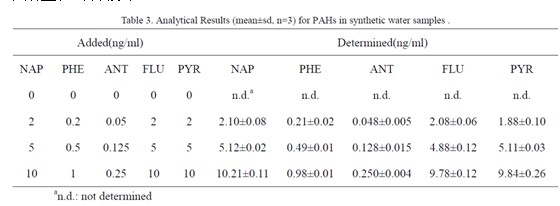
��������ʵ���������ķ���Ӧ������Ȼˮ���ķ�������ⲻ�����ܶ���ˮ�е�PAHs����˽���ӱ����ж���Һ����ȡ��HPLC ֱ�ӷ��������ý�����ڱ�4 �С�
����3.4.2 ��Ҷ��Ʒ
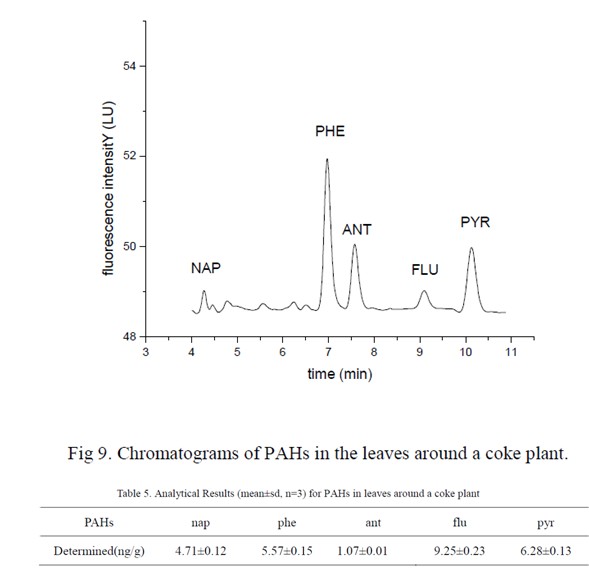
����ͼ9 �ͱ�5 �Dzɼ���ij��������������Ҷ��PAHs �ķ�����������������������������Ҷ�к���1.07-6.28 ng/g �������ƣ��죬ӫ����š�
����4 ����
������������˲��ñ��ͦ�-������ˮ��Һ��Ϊ����Һ����ȡ����ȡ�ܼ���ͨ����-�������������İ���������Ч����ȡ������Ʒ�е����ֻӷ��Զ������HPLC �ⶨ������PAHs ���·�����ʵ������������-������������Ч����߶Զ��������ȡЧ�ʣ�ͬʱ�ֶԶ������ӫ���ź��кܺõĸ������á��÷��������٣������ȸߣ�ѡ���Ժã��������л��ܼ��������ڸ��ӻ��廷��������PAHs �ķ�����
�����ο����ף�
����[1] R. Doong, S. Chang, Y. Sun, Solid-phase microextraction for determining the distribution of sixteen US Environmental Protection Agency polycyclic aromatic hydrocarbons in water samples��J. Chromatogr. A, 879(2000): 177-188.
����[2] S. Waidyanatha, Y. Zheng, et al, Determination of polycyclic aromatic hydrocarbons in urine of coke oven workers by headspace solid phase microextraction and gas chromatography�Cmass spectrometry, Chem.-Biol. Interact., 145 (2003): 165-174.
����[3] M. D. Guilen, P. Sopelana, Headspace solid-phase microextraction as a tool to estimate the contamination of smoked cheeses by polycyclic aromatic hydrocarbons, J. Dairy. Sci., 88 (2005): 13-20.
����[4] S. Shariati-Feizabad, Y. Yamini, N. Bahramifar��Headspace solvent microextraction and gas chromatographic determination of some polycyclic aromatic hydrocarbons in water samples��Anal. Chim. Acta, 489 (2003): 21-31.
����[5] O. P. Heemken, N. Theobald, B. W. Wenclawiak, Comparison of ASE and SFE with Soxhlet, Sonication, and Methanolic Saponification Extractions for the Determination of Organic Micropollutants in Marine Particulate Matter, Anal. Chem., 69 (1997): 2171-2180.
����[6] P. Navarro, E. Cortazar, L. Bartolome, et al, Comparison of solid phase extraction, saponification and gel permeation chromatography for the clean-up of microwave-assisted biological extracts in the analysis of polycyclic aromatic hydrocarbons, J. Chromatogr. A, 1128 (2006): 10-16.
����[7] P. K. Wong, J. Wang, The accumulation of polycyclic aromatic hydrocarbons in lubricating oil over time-a comparison of supercritical fluid and liquid-liquid extraction methods, Environ. Pollut. 112 (2001): 407-415.
����[8] X. Jiang, K. L. Hian, Dynamic hollow fiber-supported headspace liquid-phase microextraction, J. Chromatogr. A, 1087 (2005): 289-294.
����[9] J. B. Dallarosa, J. G. Monego, E. C. Teixeira, et al, Polycyclic aromatic hydrocarbons in atmospheric particles in the metropolitan area of Porto Alegre, Brazil, Atmos. Environ. 39 (2005): 1609-1625.
����[10] N. Saim, J. R. Dean, et al, Extraction of polycyclic aromatic hydrocarbons from contaminated soil using Soxhlet extraction, pressurised and atmospheric microwave-assisted extraction, supercritical fluid extraction and accelerated solvent extraction��J. Chromatogr. A, 791 (1997): 361-366.
����[11] L. Hou, H. K. Lee, Application of static and dynamic liquid-phase microextraction in the determination of polycyclic aromatic hydrocarbons, J. Chromatogr. A, 976 (2002): 377-385.
����[12] H. W. Chen, Determination of Polycyclic Aromatic Hydrocarbons in Water by Solid-Phase Microextraction and Liquid Chromatography, Anal. Sci., 24 (2004): 1383-1388.
����[13] L. Arthur, J. Pawliszyn, Solid phase microextraction with thermal desorption using fused silica optical fibers, Anal. Chem., 1990, 62, 2145
����[14] G. Ouyang, Y. Chen, et al, Time-Weighted Average Water Sampling with a Solid-Phase Microextraction Device, Anal. Chem. 77 (2005): 7319-7325.
����[15] H. Liu, P. K. Dasgupta, Analytical chemistry in a drop solvent extraction in a microdrop, Anal. Chem., 68 : 1817-1821.
����[16] M. A. Jeannot, F. F. Cantwell, Solvent Microextraction into a Single Drop, Anal. Chem., 68 (1996): 2236-2240.
����[17] Q. Xiao, B. Hu, Comparison of headspace and direct single-drop microextraction and headspace solid-phase microextraction for the measurement of volatile sulfur compounds in beer and beverage by gas chromatography with flame photometric detection, J. Chromatogr. A, 1125 (2006): 133-137.
����[18] A. Tankeviciute, R. Kazlauskas, Headspace extraction of alcohols into a single drop, Analyst, 126 ( 2001): 1674-1677.
����[19] C. L. Ye, Q. X. Zhou, X. M. Wang, Headspace liquid-phase microextraction using ionic liquid as extractant for the preconcentration of dichlorodiphenyltrichloroethane and its metabolites at trace levels in water samples , Anal. Chim. Acta, 572 (2006): 165-171.
����[20] S. H. Sun, Z. H. Cheng, J. P.Xie, et al, Identification of volatile basic components in tobacco by headspace liquid-phase microextraction coupled to matrix-assisted laser desorption/ionization with Fourier transform mass spectrometry, Rapid Communications in Mass Spectrometry, 19(2005): 1025-1030.
����[21] ͯ���� ��������ѧ��������Ӧ��, ��ѧ�����磬2001.
����[22] M. T. Butterfield, R. A. Agbaria, and I. M. Warner*, Extraction of Volatile PAHs from Air by Use of Solid Cyclodextrin, Anal. Chem. 68(1996): 1187-1190.
����[23] L. A. Blyshak, T. M. Rossi, G. Patonay, I. M. Warner, Cyclodextrin-modified solvent extraction for polynuclear aromatic hydrocarbons, Anal. Chem.,60(1988): 2127-2131.
����[24] N. Husain, A. Y. Christian and I. M. Warner��Effect of co-modifiers in cyclodextrin-modified mobile phases on the reversed-phase high-performance liquid chromatographic separation of polyaromatic hydrocarbons ��J. Chromatogr. A, 699(1995): 73-83.
����[25] H. Li, K. L. Hian, Application of static dynamic liquid-phase microextraction in the determination of polycyclic aromatic hydrocarbons, J. Chromatogr. A, 976(2002): 377-385.
������Ϣ
- 1������˹��ƷPractumϵ����ƽ
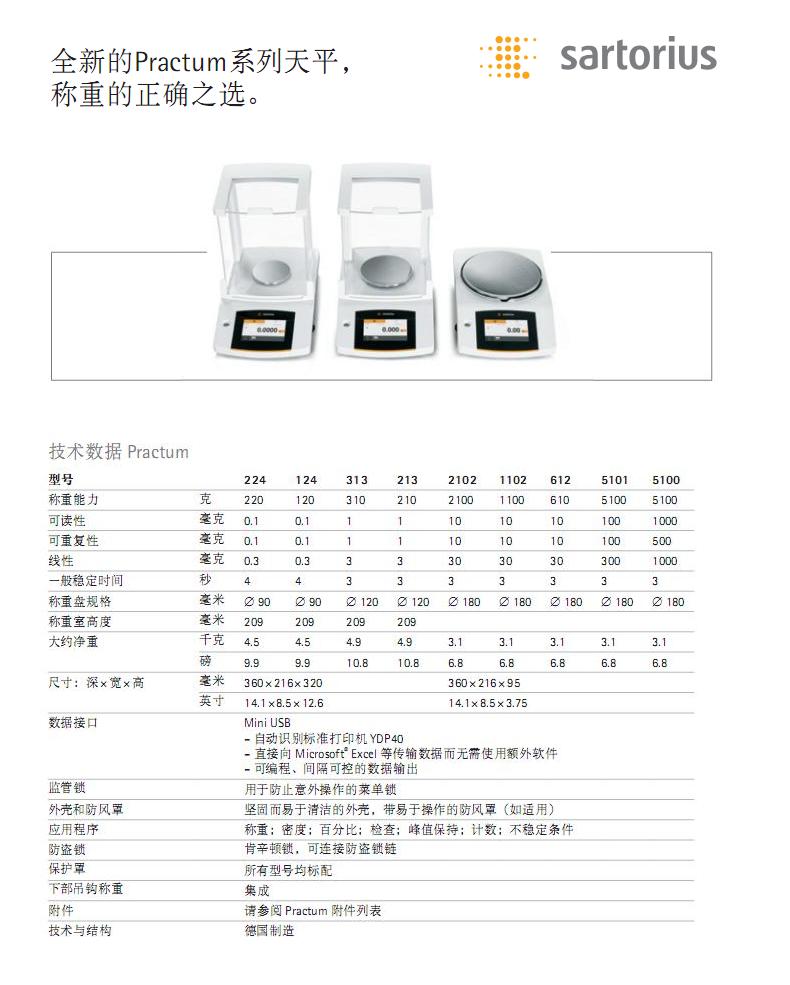
�鿴����:1283079......
- 22014���й�������ʮ��Ƽ���

���й���ѧԺ���й�����Ժ���죬�й���ѧԺԺ......
- 3�����й����������ѧ����

5��29�����磬����(�й�)��ѧ��������˾��......
- 4������ƽӦ�����ѡ�����Լ�

�鿴����:1107677 ������ƽӦ�����ѡ��......
- 5������ƽ��ʹ����ά������
������ƽ������һ������ƽ���Ǹ��ݵ����ƽ��......